Development of a Reference Database for Chemical Bonding Analysis
- Francisca Bartley
- Flash-Talks 1 , Methods
- Monday, July 14, 2025
- 12:44 pm
Francisca Bartley, Florian Kleemiss
Institute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1a, 52074 Aachen, Germany
e-mail: francisca.bartley@ac.rwth-aachen.de
The chemical bond is a fundamental concept in chemistry. However, the traditional depiction of a bond as a simple “stick” between two atoms is a crude simplification that fails to capture its quantum mechanical nature. Insights on the connection between two atoms are either derived from wavefunction-based analyses (ab initio or wavefunction fit) or from the electron densities (multipole models). Numerous techniques are available to explore different aspects of bonding. In the present work, wavefunction based methods—such as QTAIM[1], NBO[2], EDA[3], ELI-D[4], Roby Gould[5] are employed.
These bonding descriptors offer complementary perspectives hence making different assumptions depending on their depiction of the chemical bond. Some rely on the electron density, while others define the atomic boundaries using metrics like spin-dependant electron proximity, energetic contribution or orbital interaction. These different approaches may yield contradictory conclusions for the same molecule. For this reason, an unbiased interpretation requires a complementary analysis of many different descriptors for the investigated molecular structure.[6] This overarching interpretation requires reference structures to set a scale of values obtained by each descriptor. Tedious recalculations therefore hinder a wide applicability of complementary chemical bonding analysis.
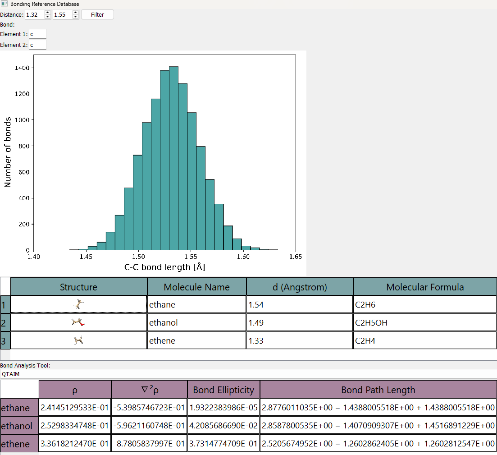
Interface for the bonding analysis database
Regardless of the chosen method, meaningful bonding analysis depends critically on the accuracy of the electron density, which can be obtained through quantum crystallography. The reliability of both quantum crystallographic results and bonding descriptors is strongly influenced by the underlying molecular geometry. Since many analyses involve gradient-based or orbital-overlap-dependent metrics, even minor distortions can shift bonding interpretations significantly.
In this context, conformational variability becomes highly relevant. While gas-phase energy minima are commonly used in theoretical studies, they may not align with geometries observed in crystal structures. An ensemble of conformers, much like polymorphism, offers a broader and more chemically relevant perspective.
Therefore, this project aims to provide a database with an intuitive user interface (see Fig. 1) of known and pre-analysed molecules along with their respective conformers. The platform enables researchers to easily compare geometries and explore bonding characteristics across multiple descriptors, fostering a more comprehensive understanding of chemical bonding.
References:
[1] R. F. W. Bader, Atoms in molecules. A quantum theory. New York: Oxford University Press 1994.
[2] C. R. Landis and F. Weinhold, The NBO View of Chemical Bonding, G. Frenking and S. Shaik (eds.), The Chemical Bond, Wiley, 2014, pp. 91-120.
[3] M. Rahm, R. Hoffmann, J. Am. Chem. Soc. 2016, 138 (11), 3731-3744.
[4] M. Kohout, Faraday discussions 2007, 135, 43-54; discussion 125-49, 503-6.
[5] K. Alhameedi, et al. Results in Chem. 2020*, 2*, 100053.
[6] S. Grabowsky (Ed.), Complementary Bonding Analysis. Berlin: De Gruyter 2021.