Implementation of density matrix tight-binding (PTB) for quantum crystallographic refinement in NoSpherA2
- Ben Ebel
- Session 10 , Methods
- Wednesday, July 16, 2025
- 11:00 am
Ben Ebel, Daniel Brüx, Florian Kleemiss
Institute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
e-mail: ben.ebel@ac.rwth-aachen.de
The computational cost associated with simulations of large or electron-rich systems, coupled with the potential for an unlimited number of combinations for the chosen level of theory, presents a significant challenge to the adoption of quantum crystallographic refinement as a standard refinement tool. A possibility to improve computational time, without sacrificing much accuracy is the use of semiempirical methods, which use empirical parameters to approximate the ab-initio approach. One method is the tight-binding (TB) approach[1], which assumes that electrons are tightly bound to the core instead of acting as a free electron gas, an assumption quite suitable for molecular crystals. Energetic contributions of a given chemical system are split into approximate contributions leading to a large decrease in computational steps. Errors from this are then accounted for through empirical parameters introduced in the method.
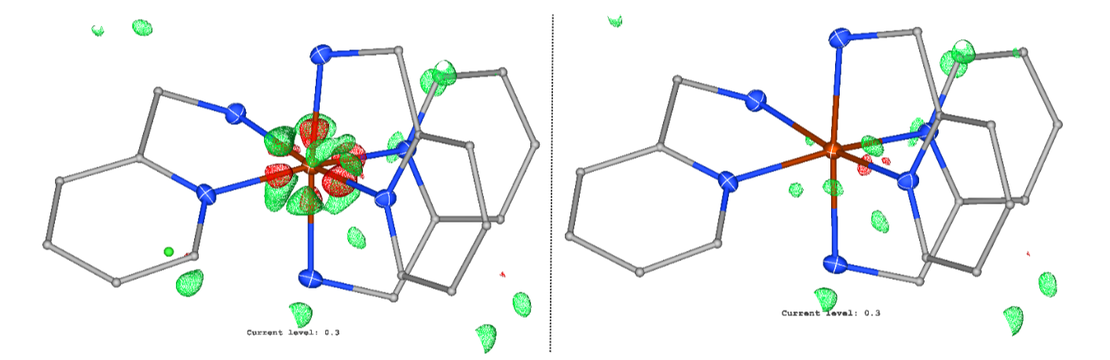
Comparison of the different methods for Co110[8] (Level: 0.330 eÅ^-3).
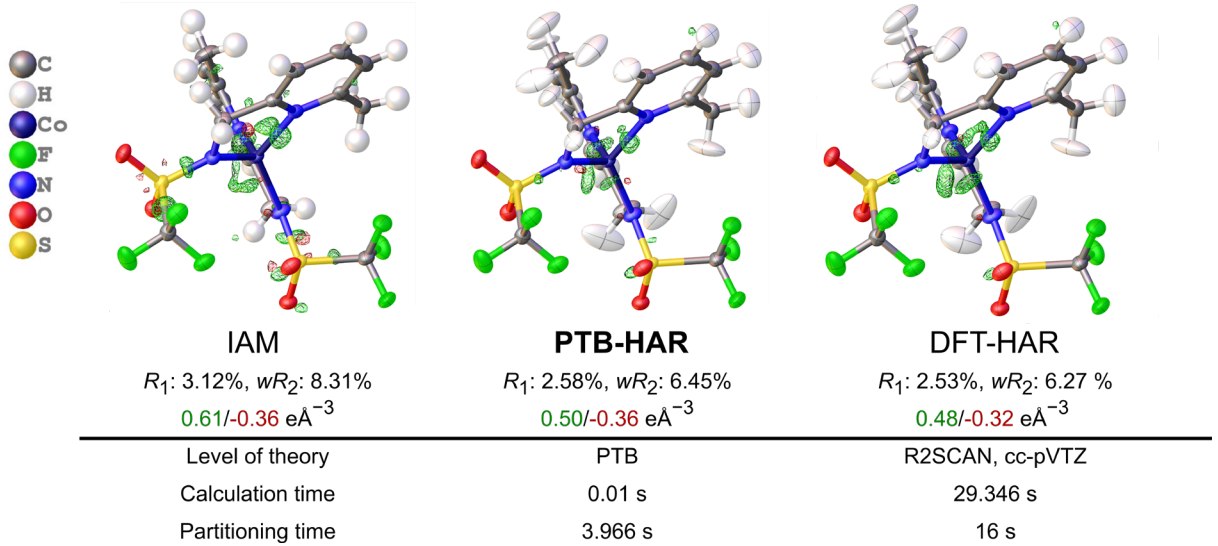
PTB[2] is a method developed by Grimme et al. to reproduce the density matrix P of the high-level range-separated hybrid density functional with an integrated double ξ basis set ωB97X-3c[3]. This matrix can then be partitioned into atomic contributions e.g. using Hirshfeld’s stockholder partition scheme to yield more accurate non-spherical atomic form factors[4–6]. We herein present the implementation of the method into NoSpherA2[7], the non-spherical refinement suite within Olex2[8] and present first results.
References:
[1] M. Elstner, G. Seifert, Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2014, 372, 20120483.
[2] S. Grimme, M. Müller, A. Hansen, J. Chem. Phys. 2023, 158, 124111.
[3] M. Müller, A. Hansen, S. Grimme, J. Chem. Phys. 2023, 158, 014103.
[4] F. L. Hirshfeld, Isr. J. Chem. 1977, 16, 198–201.
[5] D. Jayatilaka, B. Dittrich, Acta Cryst A 2008, A64, 383–393.
[6] S. C. Capelli, H.-B. Bürgi, et.al, IUCrJ 2014, 1, 361–379.
[7] F. Kleemiss, O. V. Dolomanov, et.al., Chem. Sci. 2021, 12, 1675–1692.
[8] O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, et.al., J. Appl. Cryst. 2009, 42, 339–341.