Charge Density Analysis of Organic Nanocrystals via 3D ED Data
- Anil Kumar
- Session 11
- Thursday, July 17, 2025
- 9:30 am
Anil Kumar a* Ashwin Suresh b, Arianna Lanzac, Jakub Wojciechowskid, Lukas Palatinus b* & Paulina Maria Dominiak a*
aUniversity of Warsaw, Faculty of Chemistry, Biological and Chemical Research Centre, ul. Żwirki i Wigury 101, 02-089 Warszawa, Poland, b*Institute of Physics of the Czech Academy of Sciences, Prague, Czech Republic, cUniversity of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark, dRigaku Europe SE, Neu-Isenburg, Germany
e-mail: a.kumar3@uw.edu.pl
Three-dimensional electron diffraction (3D ED) has emerged as a transformative technique in structural science, enabling crystallographic analysis of nanocrystals beyond the reach of traditional X-ray methods[1,2]. In this study, we present a detailed investigation of electron density distributions derived from 3D ED data, with emphasis on capturing subtle bonding features and charge localization. High-resolution electron diffraction data were collected for the L-alanine and urea molecules using a cryo-cooled TEM, followed by full multipole refinement using the Hansen–Coppens formalism within a dynamical scattering framework. The multipole model of experimental 3D ED data (eMMexp) was compared with the multipole models refined against theoretical (periodic DFT) static electron structure factors (eMMtheo) and high-resolution experimental X-ray diffraction data (xMMexp). Our results show strong agreement across deformation density features, electrostatic potential distributions, and topological parameters derived via Bader’s Quantum Theory of Atoms in Molecules (QTAIM). Importantly, 3D ED enabled reliable refinement of hydrogen positions and anisotropic atomic displacement parameters — surpassing conventional limitations of X-ray-based methods. This study demonstrate that 3D ED enables precise charge density mapping in organic systems, establishing it as a robust tool for quantum crystallography and the study of materials, where conventional approaches fail due to crystal size or beam sensitivity.
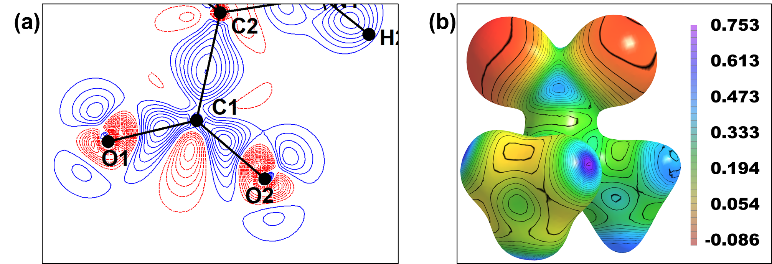
Properties computed from the eMMexp of L-alanine: (a) The 2D deformation electron density map (carboxylic group) drawn at an interval of ± 0.1 e/Å3 and (b) Molecular electrostatic potential (Bohr/Å) mapped onto the electron density isosurface of 0.05 Bohr/Å3.
Acknowledgement: The National Science Center, Poland, provided the funding for the research presented in this work under the grant 2020/39/I/ST4/02904. Acknowledgment is also extended to the Polish high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH, WCSS) for providing computer facilities and support within computational grant no. PLG/2024/017098.
References
[1] T. Gruene and E. Mugnaioli, Chem. Rev., 2021, 121, 11823–11834.
[2] M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmöller and J. P. Abrahams, ACS Cent. Sci., 2019, 5, 1315–1329.