The Segregated Atom Model for the refinement of high-resolution diffraction experiments
- Martí Gimferrer
- Session 10 , Methods
- Wednesday, July 16, 2025
- 11:30 am
Martí Gimferrer
Institut für Physikalische Chemie, Georg-August Universität Göttingen, Tammannstraße 6, 37077 Göttingen, Germany
e-mail: marti.gimferrerandres@uni-goettingen.de
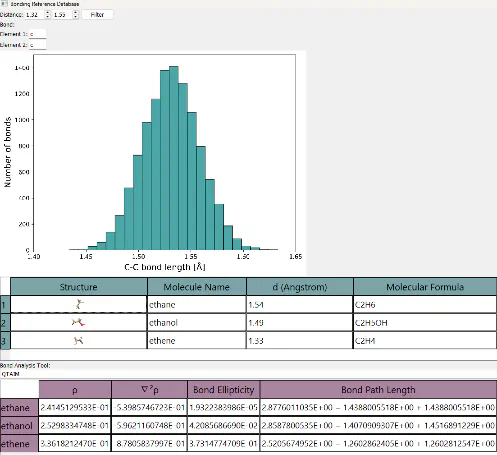
Over the last years, significant advancements in the beamlines, sample protocols, and in the data processing have pushed the field of protein crystallography towards resolutions close to or under 1Å. However, the structural refinement still relies heavily on the use of atomic form factors fitted from spherical atomic densities of isolated atoms (independent atom model, IAM), which are commonly tabulated.[1] Hence, all chemical bonding effects are ignored/lost. More sophisticated techniques, such as the multipole model (MM) or Hirshfeld atom refinements (HAR), carry the promise of solving this issue but carry some limitations as well.
We aim to include chemical bonding effects within the refinement techniques from structural biology by integrating covalent bond analysis methodologies with contemporary structure refinement techniques. For selected unrefined regions of the protein, e.g., very flexible subunits or cofactors, to name a few, we propose the on-the-fly evaluation of atomic form factors using quantum mechanical (QM) calculations, while avoiding the general framework of HAR. Specifically, the atomic densities derived from the actual QM calculation on molecules or aggregates will be employed to build a model that can be seamlessly integrated into existing crystallographic software such as SHELX or BUSTER, namely the segregated atom model (SAM). SAM factors directly depend on the atom in molecule (AIM) definition used, opening the door to tuning or developing AIM methods for crystallography based on the accuracy of the refinements. Initially, we make use of the Topological Fuzzy Voronoi Cells (TFVC)[2] as implemented in APOST-3D.[3] TFVC does not involve the use of reference densities, and it approaches QTAIM (the gold standard in the chemical bonding community) while allowing the atomic domains to overlap (smooth atomic density decay). This also allows us to address and understand the limitations of the spherical models, separating clear charge transfer from asphericity due to bonding effects. In this conference, the method implementation together with preliminary results on the AIM fine-tuning will be shown for selected benchmark systems (amino acids), compared with standard IAM refinements.
References:
[1] See for instance (most recent): Olukayode, S.; Froese Fischer, C.; Volkov, A. Revisited Relativistic Dirac–Hartree–Fock X-Ray Scattering Factors. I. Neutral Atoms with Z = 2–118. Acta Crystallographica Section A Foundations and Advances, 2023, 79, 59–79. https://doi.org/10.1107/s2053273322010944.
[2] Salvador, P.; Ramos-Cordoba, E. Communication: An Approximation to Bader’s Topological Atom. The Journal of Chemical Physics, 2013, 139. https://doi.org/10.1063/1.4818751.
[3] Salvador, P.; Ramos-Cordoba, E.; Montilla, M.; Pujal, L.; Gimferrer, M. APOST-3D: Chemical Concepts from Wavefunction Analysis. The Journal of Chemical Physics, 2024, 160. https://doi.org/10.1063/5.0206187.